发病机制
发病机制
发病机制:骨髓红细胞系显著增生导致外周血细胞容量增多的发病机制可能与下列因素有关。
1.“内生性”红细胞克隆的形成 骨髓体外进行干细胞培养时,正常骨髓细胞形成晚期红系祖细胞集落(CFU-E)需要在培养基中加EPO,而PV患者的骨髓细胞不加入EPO即能生长,提示本病患者这种不依赖EPO生成的红细胞克隆具有“肿瘤”性质。如果PV患者的骨髓培养也另外加入EPO,则在形成的CFU-E中,既有PV的细胞,又有正常红细胞,说明PV患者体内除PV细胞克隆外,尚残留正常干细胞,但其增殖受到PV克隆的抑制。目前认为PV的异常克隆是从单一细胞起源,持续增生,具有优势抑制正常克隆,同时具有细胞遗传学不稳定性,临床上可发现PV转化为急性白血病的病例。
2.红系祖细胞对EPO敏感性增强 PV患者及正常人的骨髓细胞做干细胞培养时,加入相同浓度EPO,PV患者早期红系祖细胞集落(BFU-E)和CFU-E数均比正常人显著增多,亦比患者不加EPO时生长CFU-E明显增多。当培养中加入EPO抗体后,PV患者CFU-E生成数即减少。上述结果提示PV患者红系祖细胞对EPO的敏感性增强,这也是导致红细胞增多的原因之一。
3.多能干细胞水平增殖异常 正常红细胞含A型和B型2种葡萄糖6-磷酸脱氢酶(G-6-PD)的同工酶,而PV患者的红、粒细胞和血小板仅含A型一种,而成纤维细胞和淋巴细胞中仍含A、B二型G-6-PD同工酶,说明本病是起源于同一多能干细胞水平的单一克隆性疾病。
4.细胞凋亡的异常 有研究发现PV患者有核红细胞生存时间明显长于正常人,PV集落对IL-3,SCF都高度敏感,而这些因子均能延缓红系祖细胞发生凋亡。细胞培养发现PV患者和正常对照在缺乏细胞因子的培养条件下均发生细胞凋亡,但PV患者的细胞凋亡少于正常对照,这种差异可能与PV患者bcl-2高表达有关。
5.其他 另有实验提示PV患者血清中可能有一种糖蛋白,能刺激红细胞生成,对粒细胞和血小板也有刺激作用,称为骨髓刺激因子。此因子的抗原性与:EPO不同,但需要少量EPO参与才能起作用。其性质有待进一步研究。
临床表现
临床表现:本病起病隐匿,常有数月至数年的无症状期,常在血常规检查时被发现。有的病例在出现
血栓形成和出血症状后才明确诊断。很多症状和体征与血容量和血液黏滞度增高有关。最早出现的症状常为血液循环障碍和神经系统方面的有关症状。主要临床表现有以下几个方面。
1.皮肤改变 有特征性。表现为皮肤变红,特别是颜面、颈部和肢端部位。黏膜充血,呈淡蓝色。Osler描述其症状为“夏日如玫瑰红,冬日如靛青蓝”。常见毛细血管扩张,齿龈出血和鼻衄。也见皮肤发绀、紫癜、瘀点、含铁血黄素沉积,酒渣和匙形甲。50%患者患有水源性瘙痒。可由沐浴或淋浴促发引起瘙痒,灼热或刺痒感。通常持续30~60min,与水温无关。也可发生与水无关的瘙痒。血中和皮肤中组胺增高。
2.神经系统
头痛最为常见,50%病人均有此表现,可伴头昏、
眩晕和耳鸣、疲乏、健忘、肢体麻木、多汗等。严重者可出现盲点、复视和视力模糊等视觉异常。也可有心绞痛和间歇性跛行。少数患者以脑血管意外为首发表现就诊。该组症状主要是因红细胞数增加、全血容量增多和血黏度增高而导致的血管扩张、血流缓慢淤滞和组织缺氧引起的。
3.出血 发生率<10%,主要是由于血管充血、血管内膜损伤、血小板第3因子减少等,血小板功能紊乱及凝血机制异常导致出血倾向。常见为鼻出血、牙龈出血和皮肤黏膜上的淤点和淤斑。也可表现消化道出血,拔牙后出血、
月经量多等。
4.组胺增高的表现 本症伴颗粒细胞增加,嗜碱粒细胞也增多,后者富含组胺。组胺释放增加可致消化性溃疡,故本病患者消化性溃疡发生率为10%~16%,较正常人高4~5倍,溃疡所致的上消化道大出血多见,可威胁生命。皮肤瘙痒也常见,40%发生在热水浴之后,10%可伴
荨麻疹。
5.其他 本病因骨髓细胞过度增殖,使核酸代谢过高,血液尿酸浓度升高,少数病人可发生
尿酸肾病,表现为尿结石和肾绞痛或
痛风性关节炎症状。有些病人可发生胆结石、阻塞性
黄疸和胆绞痛。最常见的体征是多血引起的面部、鼻、耳、唇、手掌和结膜充血,呈绛红色,如酒醉状。视网膜和口腔黏膜也显示充血。约70%以上患者动脉血压升高。约75%以上的患者可有脾脏肿大,通常为中、重度肿大,与
继发性红细胞增多症有一定的鉴别诊断意义。约40%患者可能有
肝大,随疾病的发展,肿大逐渐明显。
实验室检查
实验室检查:
1.红细胞
(1)
红细胞计数和
血红蛋白增高:多次检验红细胞均>6.5×10
12/L(男性)或>6.0×10
12/L(女性);
血红蛋白>180g/L(男性)或>170g/L(女性)。
(2)血细胞比容增高:男性≥54%,女性≥50%。患者常在55%~80%。
(3)用51Cr标记法测定血细胞容量大于正常值:男性>36ml/kg,女性>32ml/kg。
(4)红细胞形态改变:红细胞形态随疾病发展而变化,早期红细胞形态大多正常或轻度大小不均,当疾病发展出现脾脏高度肿大伴活跃髓外造血时,外周血出现
有核红细胞、红细胞大小、形态不等,可见椭圆、泪滴样红细胞和嗜碱点彩样红细胞。
(5)
红细胞寿命:随疾病进度而不同,病初正常或轻度缩短,晚期由于脾脏的髓外造血及单核巨噬细胞系统功能增强,
红细胞寿命可缩短。
2.粒细胞 约2/3患者白细胞计数呈中度增高,多在(12~25)×109/L,常有核左移,65%左右病人嗜碱性粒细胞绝对值增高。中性粒细胞碱性磷酸酶积分大多增高,而继发性红细胞增多患者积分一般均正常。
4.血容量及血液黏滞度 血浆容量一般正常或稍减,总血容量增多及红细胞容量增多。血液粘滞度增高,可达正常人的5~8倍。
5.骨髓象
(1)涂片几乎均显示细胞高度增生,脂肪颗粒减少,红、粒、巨核三系均增生,以红系最为显著,巨核细胞不仅数量增多而且形态增大。
(2)
铁染色显示细胞内外铁降低甚至消失,推测与慢性隐匿出血或铁利用增加、贮存铁减少有关。
(3)疾病晚期,可由于合并骨髓纤维细胞增生而呈“干抽”现象,骨髓活检比涂片检查更有助于判断骨髓纤维化的并发症,用网状纤维染色法,可以证实10%~20%的患者有纤维组织增加。
6.染色体检查 未治患者染色体异常为18%~26%,最多见的是非整倍体、假二倍体和多倍体。染色体异常大多是+8、+9及20q-。随病程延长,染色体异常发生率会逐渐升高,病期超过10年病人,染色体有异常者可达87%。PV初次诊断时,已发现有异常染色体克隆的患者生存时间比当时染色体正常者短。
7.红系祖细胞培养 PV患者的红系祖细胞在半固体培养基上可不加EPO而形成CFU-E,即内源性CFU-E,如有PV患者红系祖细胞的特点,可作为早期非典型病例的确诊依据。
8.红细胞生长素测定 应用放射免疫法测定患者血浆和尿中
红细胞生成素减少或缺如,与大多数继发性红细胞增多症有明显不同。
9.其他 绝大多数PV患者动脉
血氧饱和度在正常范围 动脉
血氧饱和度>92%,有助于除外心肺疾患引起的继发性红细胞增多症。血浆维生素B
12结合力及维生素B
12均增高 以前者更明显。此与白细胞及幼稚粒细胞释放的Ⅰ及Ⅲ型运钴胺素较多有关,这两种蛋白均能结合维生素B
12。上述两者测定有助于将本病与继发性红细胞增多症相鉴别,并可作为疗效和疾病活动指标。40%患者诊断时有高尿酸血症和高尿酸尿。60%未经治疗患者血尿中组胺升高,与血中嗜碱粒细胞增多有关。
诊断
诊断:根据皮肤改变特征性,血液细胞学检查的红细胞绝对增加,红细胞压积55%~80%。白细胞和血小板亦增多。即可诊断。1986年国际PV研究组(PVSG)制定的标准,简便易行,可供临床参考和借鉴。另外,国内根据具体情况也制定了相应的标准。
1.PVSG标准
(1)A类标准:①红细胞容量增加(
51Cr红细胞标记法):男性≥36ml/kg,女性≥32ml/kg。②动脉血氧饱和度≥0.92。③
脾大。
(2)B类标准:①血小板计数>400×109/L。②白细胞计数>12×109/L(无发热、感染状态)。③中性粒细胞碱性磷酸酶积分增高(>100,无发热、感染状态)。④血清维生素B12增高>666pmol/L或未饱和维生素B12结合力增高>1628pmol/L。
凡符合上述A类①+②+③,或A类①+②再加B类中任何2项,则可诊断。
2.国内标准 根据我国具体情况,国内制订PV诊断标准:
(1)临床表现:①皮肤、黏膜绛红色;②
脾大;③高血压或病程中有血栓史。
(2)实验室检查:
①血红蛋白及红细胞计数增加(男性血红蛋白>180g/L,红细胞>6.5×1012/L,女性分别>170g/L及6.0×1012/L)。
②血细胞容量绝对值增加,51Cr标记法红细胞容量男性>39ml/kg,女性>27ml/kg。
③血细胞比容增高,男性≥0.54,女性≥0.50。
④无感染及其他原因引起白细胞计数多次>11.0×109/L。
⑤血小板计数多次>300×109/L。
⑥外周血中性粒细胞碱性磷酸酶(NAP)积分>100。
⑦骨髓象示增生明显活跃或活跃,粒、红与巨核细胞系均增生,尤以红系细胞为显著。
诊断真性红细胞增多症可有两种方法,最好采用A法,确无条件测红细胞容量时,则采用B法。
A法:具有上述(1)类中任何2项,加(2)类中第①、②项,再加(3)类即可诊断本病。
B法:具有(1)类中第①、②项加(2)类中第①项(标准改为男性多次血红蛋白≥200g/L,女性≥190g/L),尚需具备第(2)类第③~⑦项中任何4项,再加上(3)、(4)类,方可诊断本病。
鉴别诊断
鉴别诊断:
1.继发性和
相对性红细胞增多症 继发性红细胞增多常见于下列两类情况:一是组织缺氧或肾局部缺血缺氧所致EPO分泌增加,导致红细胞代偿性增多,可见于高山病、有右至左分流的先天性心脏病、慢性肺部疾病、高铁血红蛋白症、吸烟引起的碳氧血红蛋白过多症等,患者的血氧饱和度大多降低。另一种是肾肿瘤及其他内分泌性质肿瘤自主分泌红细胞生成素或红细胞生成素样物质所致的红细胞增多症,见于肾母细胞瘤、肝癌、小脑瘤、间脑瘤、肾癌、子宫瘤等。
相对性红细胞增多症是由于血浆容量减少,使红细胞容量相对增多所致。其外周血红细胞、血红蛋白和血细胞比容增多,但全身血细胞容量正常,常见于
脱水、
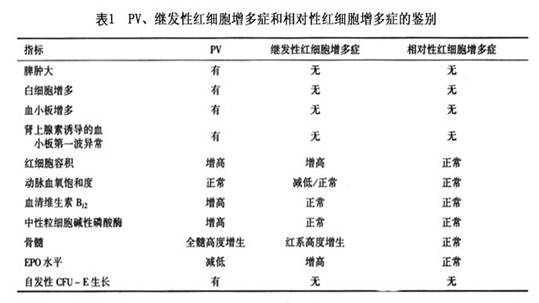
烫伤等暂时性体液丢失及因吸烟、饮酒、焦虑和高血压所致慢性相对性红细胞增多(Gaisbock综合征)。具体鉴别见表1。
2.慢性粒细胞性白血病(CML) PV患者常伴
脾大和粒细胞升高,晚期外周血幼稚粒细胞可增多,故需与CML进行鉴别。PV患者中性粒细胞碱性磷酸酶积分升高,Ph
1染色体和bcr/abl mRNA是阴性,而慢粒正好相反。近来研究发现慢粒病人也可
自发CFU-E形成,故内源性CFU-E不能用于鉴别PV和慢粒。
3.骨髓纤维化PV临床表现有许多与骨髓纤维化相似之处 PV晚期也可继发骨髓纤维化,两者主要鉴别是病史和骨髓活检,骨髓纤维化骨髓病理示纤维组织明显增多,而PV主要表现为髓外造血现象,只有晚期才合并骨髓纤维化,且病变范围小,程度较轻。
治疗
治疗:抑制骨髓红系细胞异常增生、降低血容量、减少血黏度、消除红细胞增多所致的各种症状和体征、减少血栓栓塞及出血性并发症、提高生活质量并延长生存期是治疗PV的目标。
1.静脉放血 每周静脉放血2~3次,每次400ml,直至HCT正常。此种治疗手段常可迅速缓解症状及降低红细胞容量,但不能使升高的白细胞和血小板下降也不能缓解顽固的皮肤瘙痒及痛风发作。有心、脑血管病或有血栓史者,放血宜慎重,每次以250ml为好,每周至多2次,目标为HCT维持于42%~45%。为防止血栓形成,放血后可静脉输注
右旋糖酐40(
低分子右旋糖酐)500ml。反复放血者可致缺铁,需适当补充之。
我国由于传统习惯的原因,放血疗法始终难以广泛开展,尤其是每周均需放血更不易被接受。因此,做好宣传解释工作是放血疗法的重要组成部分。尤其应强调其发生白血病转化(仅1.5%)及继发实体瘤的比例最低,以及不良反应最少,且中数生存期和其他疗法相近,为12.6年。但单独放血疗法者前3年的血栓栓塞性并发症发生率较高,此后伴发骨髓纤维化者也较多。必须强调,即使单独放血治疗者,其发生白血病转化较其他疗法为低,但仍明显高于相匹配的正常人群。目前较一致的看法是,病情稳定的年轻患者较适合行放血治疗,并辅以低剂量阿司匹林治疗。
2.骨髓抑制性治疗
(1)放射性核素治疗:32P使用最多,其通过释放β射线阻止骨髓造血细胞的核分裂,从而抑制造血。经首剂静脉注射2~3mci/m2后,多数病例在4~8周内血象恢复正常。如3个月后血象未能纠正者,可第2次给药,剂量宜增加25%。少数患者需第3次给药,但1年内总剂量不应>15mci。32P也可口服给药,但剂量应增加25%,分2次,间隔1周给予。32P治疗的缓解率可达75%~85%,疗效可持续半年至数年,并可降低血栓栓塞性并发症发生率。其缺点为,如剂量掌握不当,过大可造成骨髓抑制。其次为治疗后急性白血病及实体瘤的发生率明显高于静脉放血者,尤其是远期急性白血病的发生率高达10.3%。如32P治疗后再并用化疗者,急性白血病的发生率更高。鉴于上述原因,目前32P主要用于老年患者。32P治疗者的中数生存期为10.9年。
(2)化学药物治疗:
①
羟基脲(
Hydroxyurea,
HU):
HU在欧美应用最普遍,剂量为1.5~2g/d,几周内血象可达正常范围,再以0.5~1.0g/d维持。
HU疗效短暂,停药后常迅速反跳,故需持续用药。一旦发生骨髓抑制,在停药后数天至数周即可恢复。长期
HU治疗者,5.4%发生急性白血病,虽然仍高于静脉放血者,但安全性相对较好。
HU发生骨髓纤维化及死亡率和静脉放血者相似,而血栓栓塞性并发症则明显降低,仅6%。
②烷化剂:
白消安(
Busulfan,myeleukon)在国内应用最多,剂量为4~6mg/d。通常用药1月左右才能控制血象,但作用持续时间明显长于
HU,因此可间断给药。部分病例停药后数月,甚至数年血象仍维持基本正常,中数缓解期可达4年。间断用药可减低远期急性白血病的发生率,有报道仅2%。另一烷化剂苯丁酸氮芥(Chlorambucil,CB
l348)作用较
白消安弱而慢,中数生存期为9.1年。其急性白血病的转化达17%,另有3.5%的患者并发大细胞淋巴瘤,故目前已较少应用。
③
三尖杉碱类:为我国首创的抗白血病药物,对急、慢性髓细胞白血病均有效,20世纪80年代应用于PV后发现有较好的疗效。此类药物包括
三尖杉碱和
高三尖杉酯碱,剂量均为2mg/d,静脉滴注或肌内注射,10~14天为1疗程。一般在停药后1~2个月血象降至正常,疗效大多维持3~6个月,少数可维持1年以上。复发后再次用药通常仍有效。按上述剂量及疗程用药,绝大多数患者不发生骨髓抑制,心脏毒性也少见。远期是否会促使转化为白血病,尚无确切资料。另有报告,采用每天2~4mg静脉滴注,连续或间歇用至红细胞及血红蛋白正常,可延长缓解期达10个月以上,但部分病人可伴白细胞和(或)血小板减少。
3.本病伴发的瘙痒治疗困难。可用抗
组胺药物如盐酸赛庚啶单独或与西咪替丁联合应用。其他如阿司匹林和PUVA亦可应用。
4.其他治疗 近年有报道应用基因重组的干扰素α(IFN-α)治疗PV,已取得一定疗效。其抑制异常克隆的造血祖细胞及骨髓成纤维细胞的增殖,拮抗血小板衍生生长因子(PDGF)及转移生长因子(TGF-β),以减轻骨髓纤维化。由于IFNα起效慢,故宜在应用其他治疗,待血象明显好转后再用,作为长期维持治疗。IFNα的剂量为300万~500万U/次,每周3次,疗程至少6~12个月。单用IFNα的反应率为60%。有报道低剂量阿司匹林(50mg/d)即可使血栓素A2的产生减少80%以上,故推荐长期应用,尤其适用于单独静脉放血治疗者,以减少血栓栓塞性并发症。但有应用后引起出血的报道,而反映血小板功能的实验室检测方法并不能预报出血的危险。因此,建议用250mg/d以下的剂量,以往有出血史者禁用。
各种抗
组胺药物对PV患者的顽固性瘙痒效果欠佳,已有报道IFNα有一定效果,但起效较慢。骨髓抑制性治疗控制血象后,才能获缓解。PV晚期合并骨髓纤维化(有人称之为PV的衰竭期),患者常有巨脾、贫血、白细胞、血小板减少,处理十分困难。脾区放疗已证实无效,脾切除至少可取得暂时的缓解。由于手术并发症多,病死率高达25%,应谨慎进行,并术前做好充分的准备。重度贫血者常需定期输血,也可使用雄性激素。缺铁时补充铁剂宜慎重,因可促使红细胞短期迅速增加而加重病情。
PV患者因并发外科疾病的手术,包括拔牙,术后并发症高达47%,其中大多为出血,或血栓性并发症,风险较大。故主张术前先行放血及血细胞置换,待血象明显好转后再手术。
预后
预后:PV大多发展缓慢,未经治疗者的中数寿命为1.5年,但经各种治疗后,中数生存期可达10~15年。
PV在病程中可发生各种转化,北京协和医院随访病程3年以上的PV 90例,在病程中10例转化为其他的骨髓增殖性疾病和(或)急性白血病,转化率为16.7%。部分病例可有多种转化,如先转为血小板增多症(此时红细胞数及容量均正常),后再转化为骨髓纤维化,最终转为急性白血病。此外,个别病例可转化为慢性淋巴细胞白血病。文献中较多作者提出,PV转化为骨髓纤维化后,20%~50%将进展为急性白血病,其中绝大多数为急性髓性白血病。PV可直接转化为急性白血病,也可经骨髓增生异常综合征(MDS)阶段再转化为急性白血病,二者均各占50%。一旦转化为急性白血病,各种治疗效果均差,通常在数月内死亡。
PV的首位死因是血栓栓塞性并发症,占30%~40%,其中心肌梗死占50%、脑卒中占31.5%、
静脉血栓占18.5%。其他依次为急性白血病(19%)、实体瘤(5%)、出血(5%)。余下的病例死于晚期骨髓衰竭(包括骨髓纤维化),其中大多数因中性粒细胞缺乏,死于感染,另为血小板减少,死于内脏出血。